: _- n: | E+ N8 I+ Q" u1 _) S1 Yе®ӢеҗҜж–Ңж•ҷжҺҲпјҡз”ҹзү©ж Үеҝ—дёҺйқһе°Ҹз»ҶиғһиӮәзҷҢзҡ„дёӘдҪ“еҢ–иҜҠж–ӯе’ҢжІ»з–—
! C4 {+ V) e' S7 \% D1 i0 [
1 F% Y. M; u4 R/ S2014-06-08 03:28 жқҘжәҗпјҡдёҒйҰҷеӣӯ 9 N1 H% r( R7 Q8 d3 S
еӯ—дҪ“еӨ§е°Ҹпјҡ ) ]* s- ~& Y4 o) f P! k- f6 Z9 A- {' f3 O
% ~* I$ }7 u' @+ N
, e3 `9 g' _: D/ X; k' e# g9 w
, |) J1 B# k6 ^жӯҰжұүеӨ§еӯҰдәәж°‘еҢ»йҷўиӮҝзҳӨдёӯеҝғ е®ӢеҗҜж–Ң иғЎ иғң
% R- K4 Z* f( J
0 S1 c& `4 H/ w9 z+ y6 h
. O$ `. Z Y; Nе®ӢеҗҜж–Ңж•ҷжҺҲеӣ дәӢжңӘеҲ°еңәпјҢз”ұиғЎиғңд»ЈдёәжҠҘе‘Ҡ
7 I% E% \; x, j( h7 N6 {: j; R5 [3 g
, G0 ^1 A# W1 \( X5 A, c4 R& k n/ V$ o, f g
PPTд№ӢдёҖ, B" T( Y3 Q$ h+ a. F* b- M
* x2 J1 {; D* e; z! V+ O8 w6 T. b7 ^) r: L& m. N% b3 B
PPTд№ӢдәҢ
9 n2 {1 M' `% ~1 X- q; S" E* X3 t& B! Z
ж‘ҳиҰҒ TNMеҲҶжңҹзі»з»ҹиҷҪ然еңЁжІ»з–—йҖүжӢ©гҖҒйў„еҗҺеҲӨж–ӯдёӯеҸ‘жҢҘйҮҚиҰҒдҪңз”ЁпјҢдҪҶ并дёҚжҳҜзҷҢз—Ү(еҢ…жӢ¬NSCLC)жІ»з–—зӯ–з•Ҙзҡ„е®ҢзҫҺж ҮеҮҶгҖӮдҪҝз”Ёеҹәеӣ з»„гҖҒиӣӢзҷҪз»„жҠҖжңҜпјҢе·Із»ҸеҸ‘зҺ°дәҶеӨҡз§Қе…ій”®еҹәеӣ гҖҒзӘҒеҸҳ(еҰӮEGRF)е’Ңз»ҶиғһеҶ…дҝЎеҸ·еҲҶеӯҗгҖҒйҖҡи·ҜвҖ”вҖ”з”ҹзү©ж Үеҝ—пјҢеҸҜиғҪдёҺиӮәзҷҢзҡ„еҲҶжңҹгҖҒйў„еҗҺе’ҢиҚҜзү©ж•Ҹж„ҹжҖ§жңүе…іпјҢдҪҶдёҙеәҠдҪҝз”Ёзҡ„д»·еҖјзӣ®еүҚиҝҳжңӘеҸ–еҫ—е®Ңе…ЁдёҖиҮҙгҖӮиҒ”еҗҲеӨҡдёӘз”ҹзү©ж Үеҝ—пјҢз”ҡиҮід»Ҙж•ҙдёӘдҝЎеҸ·йҖҡи·Ҝдёәж Үеҝ—пјҢдјҳеҢ–NSCLCдёӘдҪ“еҢ–жІ»з–—иҝӣиЎҢзҡ„ж—¶д»Је°ҶдјҡеҲ°жқҘгҖӮ
7 C! E# C4 B2 h( n1 W/ L
* e; z0 h5 q& i дёҖгҖҒ иӮәзҷҢзҡ„жөҒиЎҢз—…еӯҰ9 ~6 Y e# a2 R3 {$ y
: i! Z* q. E! r) B еңЁдё–з•ҢиҢғеӣҙеҶ…пјҢиӮәзҷҢзҡ„еҸ‘з”ҹзҺҮеұ…жҒ¶жҖ§иӮҝзҳӨ第дәҢдҪҚпјҢдҪҶдёәжҒ¶жҖ§иӮҝзҳӨжӯ»дәЎзҡ„йҰ–иҰҒеҺҹеӣ гҖӮ2002е№ҙе…Ёдё–з•ҢиӮәзҷҢзҡ„ж–°еҸ‘з—…дҫӢдёә130дёҮпјҢеҮ д№ҺдёҖеҚҠ(49.9%)еҸ‘з”ҹеңЁеҸ‘еұ•дёӯеӣҪ家гҖӮдј°и®ЎеҲ°2020е№ҙжҜҸе№ҙж–°иҜҠж–ӯзҡ„иӮәзҷҢжӮЈиҖ…д»ҚдјҡжҢҒз»ӯеҚҮй«ҳгҖӮ2007е№ҙзҫҺеӣҪж–°иҜҠж–ӯзҡ„иӮәзҷҢжӮЈиҖ…йў„и®Ўдёә213 380пјҢжӯ»дәЎжӮЈиҖ…дёә160 390гҖӮжҲ‘еӣҪ2000е№ҙиӮәзҷҢз”·жҖ§и°ғж•ҙеҸ‘з—…зҺҮдёә38.5/10дёҮпјҢжӯ»дәЎзҺҮ33.2/10дёҮпјӣеҘіжҖ§и°ғж•ҙеҸ‘з—…зҺҮдёә15.7/10дёҮпјҢжӯ»дәЎзҺҮдёә13. 5/10дёҮпјӣ2000е№ҙеҲ°2005е№ҙпјҢиӮәзҷҢзҡ„еҸ‘з”ҹзҺҮдёҠеҚҮдәҶ30.5%гҖӮйқһе°Ҹз»ҶиғһиӮәзҷҢ(non-small cell lung cancerпјҢNSCLC)зәҰеҚ иӮәзҷҢзҡ„80%пјҢе…¶з–—ж•Ҳд»ӨдәәеӨұжңӣпјҢзҫҺеӣҪ5е№ҙз”ҹеӯҳзҺҮдёә15%пјҢ欧жҙІдёә10%пјҢеҸ‘еұ•дёӯеӣҪ家з”ҡиҮідҪҺдәҺ8.9%гҖӮеӨҚеҸ‘е’ҢйҮҚиҰҒеҷЁе®ҳзҡ„иҪ¬з§»жҳҜжІ»з–—еӨұиҙҘзҡ„дё»иҰҒеҺҹеӣ гҖӮ5 u( S( `) k6 H7 {; L$ Y: H
! [8 _' T8 F; z' G4 p: F; f9 M дәҢгҖҒ NSCLCдёӘдҪ“еҢ–жІ»з–—зҡ„зҺ°зҠ¶
7 u8 r! X# n- }
: f+ m& \ {/ Q( } зҷҢз—ҮдёӘдҪ“еҢ–жІ»з–—жҳҜжҢҮж №жҚ®жҜҸдёӘжӮЈиҖ…зҡ„дёҚеҗҢз—…жғ…пјҢеҲ¶е®ҡзү№ејӮзҡ„жІ»з–—ж–№жЎҲпјҢз»ҷдәҲжӮЈиҖ…жңҖйҖӮеҪ“жңүж•Ҳзҡ„жІ»з–—пјҢдҪҶзӣ®еүҚзҡ„NSCLCдёӘдҪ“еҢ–жІ»з–—иҝҳд»…еҒңз•ҷеңЁTNMеҲҶжңҹзҡ„еұӮж¬ЎдёҠгҖӮ
2 ], E& q) V! V; M' o$ ?0 V
0 e) `0 ~% s3 y- y) @ TNM еҲҶжңҹзі»з»ҹз”ұAJCC(American Joint Committeeon Cancer)дәҺ1958е№ҙйўҒеёғпјҢеҮ д№Һдё“й—ЁдҫқжҚ®з–ҫз—…зҡ„и§Јеү–еӯҰиҢғеӣҙеҲ¶е®ҡгҖӮе…¶дё»иҰҒеҠҹиғҪжҳҜжҸҗдҫӣйў„еҗҺдҝЎжҒҜпјҢйҖүжӢ©еҲқе§ӢжІ»з–—зӯ–з•Ҙпјӣе…¶ж¬ЎжҳҜеңЁдёҙеәҠиҜ•йӘҢдёӯеҲҶеұӮжӮЈиҖ…пјҢдҪҝеҢ»еӯҰдёӯеҝғд№Ӣй—ҙзҡ„з ”з©¶з»“жһңдәӨжөҒжӣҙеҮҶзЎ®гҖӮеҒ¶е°”пјҢиӢҘAJCCи®ӨдёәиӮҝзҳӨзҡ„еҲҶзә§гҖҒз»ҶиғһеӯҰдәҡеһӢжҲ–жӮЈиҖ…зҡ„е№ҙйҫ„еҸҜд»ҘжҸҗй«ҳйў„и®Ўз”ҹеӯҳзҡ„еҮҶзЎ®жҖ§жҲ–жІ»з–—еҸҚеә”пјҢд№ҹдјҡиў«еҠ е…ҘеҲ°TNMзі»з»ҹгҖӮ8 f: J! a+ F* k
$ J! H; B' y4 W# n дҪҶTNMеҲҶжңҹзі»з»ҹ并дёҚиғҪи§ЈйҮҠеҗҢж ·зҡ„еҲҶжңҹе’ҢжІ»з–—ж–№жЎҲпјҢжӮЈиҖ…е…¶йў„еҗҺе·®ејӮеҫҲеӨ§з”ҡиҮіжҲӘ然дёҚеҗҢзҡ„зҺ°иұЎгҖӮжҸҗзӨәTNMеҲҶжңҹдёҚжҳҜйҖүжӢ©зҷҢз—Ү(еҢ…жӢ¬NSCLC)жІ»з–—зҡ„е®ҢзҫҺж ҮеҮҶгҖӮ
9 K7 ?* }4 I8 t; N- _8 S
Q: {; W) U9 L2 ]6 D9 w) j еәҶе№ёзҡ„жҳҜпјҢзӣ®еүҚеҜ№дәҺйў„еҗҺеҲӨж–ӯе’ҢйҖүжӢ©жІ»з–—ж–№жі•пјҢеҮәзҺ°дәҶж–°зҡ„жңәдјҡгҖӮйҰ–е…ҲпјҢдҫқжҚ®еҹәеӣ иЎЁиҫҫж Үеҝ—зҡ„е·®ејӮеҸҜе°ҶNSCLCеҲҶдёәйў„еҗҺжңүжҳҺжҳҫе·®ејӮзҡ„дәҡеһӢ(зӢ¬з«ӢдәҺдј з»ҹзҡ„з—…зҗҶеӯҰеҲҶзұ»)пјҢд»ҺиҖҢеңЁеҲҶеӯҗж°ҙе№ідёҠжҢҮеҜјиӮәзҷҢзҡ„дёӘдҪ“еҢ–жІ»з–—гҖӮ
$ u' B0 C) B1 D: Y$ J7 [
- J8 Q$ R1 V" \/ I9 I; W 第дәҢпјҢдёҺTNMжҸҗеҮәзҡ„ж—¶д»ЈдёҚеҗҢпјҢж–°еһӢеҢ–з–—иҚҜзү©жӣҙжңүж•Ҳе’Ңжӣҙе№ҝжіӣзҡ„дҪҝз”ЁпјҢеҰӮзҙ«жқүйҶҮгҖҒеҗүиҘҝд»–ж»ЁгҖҒй•ҝжҳҘз‘һж»ЁгҖҒеҹ№зҫҺжӣІиөӣгҖӮ' g$ b0 g% O8 Y. E# l, Z
6 a2 d0 x& O9 i% Y$ ?3 d0 t4 R2 Y
第дёүпјҢж–°зҡ„йқ¶еҗ‘иҚҜзү©пјҢеҰӮErlotinib (Tarceva)пјҢGefitinib (Iressa)е’ҢHeceptinпјҢд»…д»…еҜ№е…·жңүзү№ејӮеҲҶеӯҗж Үеҝ—зҡ„жӮЈиҖ…жңүж•ҲгҖӮеҰӮGefitinibз”ЁдәҺиӮәзҷҢзҡ„жІ»з–—пјҢз–—ж•Ҳз”ұEGFRеҹәеӣ зӘҒеҸҳгҖҒжӢ·иҙқж•°зӣ®жүҖеҶіе®ҡпјҢжҸҗзӨәEGFRеҹәеӣ зҠ¶жҖҒд№ҹжҳҜдёҖз§ҚеҲҶеӯҗз”ҹзү©ж Үеҝ—гҖӮ i8 ?% V5 A# L
) C3 y8 d V2 s8 J# Q- M/ L6 G
иҷҪ然зӣ®еүҚз”ҹзү©ж Үеҝ—иҝҳжІЎжңүжӯЈејҸеҠ е…ҘNSCLCзҡ„TNMеҲҶжңҹзі»з»ҹпјҢдҪҶж–°еҸ‘зҺ°зҡ„иӮәзҷҢз”ҹзү©ж Үеҝ—пјҢз”ЁдәҺNSCLCдёӘдҪ“еҢ–жІ»з–—зҡ„з ”з©¶е·Із»ҸеҸ–еҫ—дәҶйҮҚеӨ§иҝӣеұ•гҖӮеҸҜд»Ҙйў„и®ЎпјҢз”ҹзү©ж Үеҝ—жӯЈејҸеҠ е…ҘеҲ°TNMеҲҶжңҹзі»з»ҹпјҢжҲ–дҪңдёәдёҖдёӘиЎҘе……иҝӣиЎҢеҚҸеҗҢиҜ„дј°(co-evaluation)зҡ„ж—¶д»ЈеҚіе°ҶеҲ°жқҘгҖӮ& Q ^$ P0 b. c2 X) O0 f
7 U' R* E+ @3 {! ]8 }1 m8 J дёүгҖҒ NSCLCдёӯзҡ„з”ҹзү©ж Үеҝ—
: F' e6 H& j% _' A" |' I( r* f3 [5 q4 {9 d0 R4 i
жҒ¶жҖ§з»Ҷиғһзҡ„еўһж®–е’ҢиҪ¬з§»дёҖиҲ¬з”ұеҺҹзҷҢеҹәеӣ жҝҖжҙ»гҖҒиӮҝзҳӨжҠ‘еҲ¶еҹәеӣ зҡ„еӨұжҙ»е’Ң/жҲ–DNAдҝ®еӨҚжңәеҲ¶еӨұжҙ»еҜјиҮҙпјҢд»ҘеҸҠе…¶д»–ејӮеёёпјҢеҰӮRNAзҡ„иЎЁиҫҫгҖҒиӣӢзҷҪзҡ„зҝ»иҜ‘е’Ңзҝ»иҜ‘еҗҺзҡ„дҝ®йҘ°е’ҢеҠҹиғҪгҖӮ, ^2 A) u3 J5 v) X1 \; D, m/ Z
/ g6 {& l' D& h
еӣ жӯӨпјҢDNAеҹәзЎҖзҡ„з”ҹзү©ж Үеҝ—еҢ…жӢ¬SNP(еҚ•ж ёиӢ·й…ёеӨҡжҖҒжҖ§)гҖҒжҹ“иүІдҪ“ејӮеёёгҖҒDNAжӢ·иҙқж•°зӣ®ејӮеёёд»ҘеҸҠеҗ„з§ҚеҗҜеҠЁеӯҗеҢәеҹҹзҡ„з”ІеҹәеҢ–гҖӮRNAзҡ„з”ҹзү©ж Үеҝ—еҢ…жӢ¬пјҢеҹәеӣ иҝҮиЎЁиҫҫжҲ–дҪҺиЎЁиҫҫпјҢеҗ„з§Қи°ғиҠӮRNA(еҰӮе°ҸRNAгҖҒеҫ®RNA)гҖӮиӣӢзҷҪз”ҹзү©ж Үеҝ—еҢ…жӢ¬з»ҶиғһиҶңиЎЁйқўеҲҶеӯҗгҖҒиӮҝзҳӨжҠ—еҺҹгҖҒиӣӢзҷҪзЈ·й…ёеҢ–гҖҒзі–еҹәеҢ–зҠ¶жҖҒгҖӮ( k2 p! i. }* ]0 a
6 Z' O; Q# x1 G0 O7 I еҪ“然пјҢиӮәзҷҢзҡ„иӮҝзҳӨз”ҹзү©ж Үеҝ—д№ҹеҸҜеҲҶдёәиЎҖжё…з”ҹзү©ж Үеҝ—гҖҒз»„з»Үз”ҹзү©ж Үеҝ—гҖҒеҲҶжіҢзү©(е”ҫж¶ІгҖҒз—°ж¶І)з”ҹзү©ж Үеҝ—гҖӮиЎҖжё…з”ҹзү©ж Үеҝ—жңҖе…·еҗёеј•еҠӣпјҢеӣ дёәе…¶еңЁд»»дҪ•ж—¶еҖҷеқҮе®№жҳ“иҺ·еҫ—пјӣеҗ«жңүеӨ§йҮҸзҡ„з”ҹзү©дҝЎжҒҜ(еҰӮдҪҺеҲҶеӯҗйҮҸзҡ„иЎҖжё…еӨҡиӮҪз»„пјҢPeptidome)гҖӮNSCLCзҡ„з”ҹзү©ж Үеҝ—йҷӨдәҶеёёи§Ғзҡ„CEAгҖҒNSEеӨ–пјҢиҝҳеҢ…жӢ¬з”ҹй•ҝеӣ еӯҗеҸҠе…¶еҸ—дҪ“гҖҒзҷҢеҹәеӣ пјҢеҰӮK-rasгҖҒEGFRе’ҢHER-2гҖӮжӯӨеӨ–пјҢиҝҳжңүз»Ҷиғһе‘Ёжңҹзү№ејӮиӣӢзҷҪе’ҢеҮӢдәЎи°ғиҠӮеҲҶеӯҗпјҢеҰӮp53гҖҒBcl2гҖҒRbе’Ңp16INK4aгҖӮе…¶д»–еҢ…жӢ¬DNAдҝ®еӨҚзӣёе…іеҲҶеӯҗгҖҒVEGFгҖҒе№Із»Ҷиғһеӣ еӯҗ(stem cell fatorпјҢSCF)е’ҢиӮқз»Ҷиғһз”ҹй•ҝеӣ еӯҗ(HGF)гҖӮиҒ”еҗҲеӨҡдёӘж Үеҝ—жҜ”еҚ•дёӘж Үеҝ—жӣҙжңүйҖүжӢ©жҖ§е’Ңжңүж•ҲжҖ§гҖӮ) G- Q( s1 S, M
е°Ҫз®Ўе·ІеҸ‘зҺ°з”ҹзү©ж Үеҝ—еҸҜиғҪдёҺиӮәзҷҢзҡ„еҲҶжңҹжҲ–йў„еҗҺжңүе…іпјҢдҪҶзӣ®еүҚзҡ„дёҙеәҠдҪҝз”Ёзҡ„д»·еҖјиҝҳжңӘеҸ–еҫ—дёҖиҮҙгҖӮдҪҶжҳҜпјҢеҸҚеә”дёҖдәӣе…ій”®з»ҶиғһеҶ…дҝЎеҸ·йҖҡи·Ҝзҡ„з”ҹзү©ж Үеҝ—зҡ„з ”з©¶е·Із»ҸеҸ–еҫ—зӣёеҪ“еӨ§зҡ„иҝӣеұ•пјҢе°Өе…¶зү№ејӮзҡ„еҸ—дҪ“й…Әж°Ёй…ёжҝҖй…¶дҝЎеҸ·йҖҡи·Ҝ(EGFR)гҖӮ
' t3 u k0 a" t# c- Z+ G& c' U! `. f
9 F- h- M. K/ q& m* R5 p, v еӣӣгҖҒ з”ҹзү©ж Үеҝ—зү©дёҺNSCLCзҡ„иҜҠж–ӯеҲҶзұ». d5 `, G6 L% s, h7 c6 ^
( f ~7 w1 v9 J: s% @ еңЁиӮәзҷҢдёӯпјҢзӣ®еүҚдёҖдёӘйҮҚиҰҒзҡ„ж–№еҗ‘жҳҜдҪҝз”Ёеҹәеӣ е’ҢиӣӢзҷҪеҫ®йҳөеҲ—(иҠҜзүҮ)жҺўи®ЁзҷҢз—Үз»Ҷиғһзҡ„еҲҶеӯҗз”ҹзү©еӯҰзү№жҖ§гҖӮSugitaзӯүз”Ёеҹәеӣ иҠҜзүҮе’Ңз»„з»ҮиҠҜзүҮжЈҖжөӢиӮәзҷҢз»Ҷиғһзі»е’Ң187дҫӢдёҙеәҠNSCLCж Үжң¬пјҢеҸ‘зҺ°зҷҢз—Ү/зқҫдёёжҠ—еҺҹ(Cancer/Testis AntigensпјҢCTAg)еҸҜд»ҘдҪңдёәдёӯеӨ®еһӢиӮәйіһзҷҢе’Ңе°Ҹз»ҶиғһиӮәзҷҢзҡ„з”ҹзү©ж Үеҝ—зү©пјҢз”ЁдәҺж—©жңҹиҜҠж–ӯе’Ңз–—ж•Ҳзӣ‘жөӢгҖӮInamuraзӯүеҜ№48дҫӢйіһзҷҢдҪҝз”Ё40 386еҹәеӣ иҝӣиЎҢзӯӣжҹҘпјҢеҸ‘зҺ°91дёӘдёҺз»ҶиғһеҲҶеҢ–жңүе…ізҡ„еҹәеӣ еңЁйў„еҗҺеҘҪзҡ„з»„й«ҳиЎЁиҫҫпјҢиҖҢ1 499дёӘдёҺзҝ»иҜ‘еҠҹиғҪзӣёе…ізҡ„еҹәеӣ еңЁйў„еҗҺе·®зҡ„з»„й«ҳиЎЁиҫҫгҖӮ p1 i- w! w& ]% U2 ]
8 V* g- p! y9 h7 A" L
з”ҹзү©ж Үеҝ—зү©з”ЁдәҺиӮәзҷҢеҲҶзұ»з ”究зҡ„жңҖеӨҡзҡ„жҳҜиӮәи…әзҷҢ(и…әзҷҢзҡ„еҸ‘з”ҹзҺҮжңҖй«ҳ)гҖӮ2001е№ҙпјҢBhattacharjeeзӯүйҰ–ж¬ЎдҪҝз”Ёеҫ®йҳөеҲ—ж–№жі•еҜ№186дҫӢиӮәзҷҢ(127дҫӢи…әзҷҢ)е’Ң17жӯЈеёёиӮәз»„з»ҮиҝӣиЎҢеҲҶжһҗпјҢеҸ‘зҺ°и…әзҷҢеҸҜд»ҘеҶҚеҲҶдёә4зұ»(C1гҖҒC2гҖҒC3е’ҢC4)пјҢC1дё»иҰҒиЎЁиҫҫеўһж®–зӣёе…ізҡ„еҹәеӣ пјҢC2зҘһз»ҸеҶ…еҲҶжіҢзӣёе…ізҡ„еҹәеӣ иЎЁиҫҫпјҢC3дё»иҰҒиЎЁиҫҫйёҹж°Ёй…ёи„ұзҫ§й…¶е’Ңи°·иғұз”ҳиӮҪ-S-иҪ¬з§»й…¶еҹәеӣ пјҢC4дё»иҰҒиЎЁиҫҫв…ЎеһӢиӮәжіЎдёҠзҡ®ж Үеҝ—гҖӮиҝҷз§ҚеҲҶзұ»дёҺдёҙеәҠз—…зҗҶд№ҹеӯҳеңЁиҒ”зі»пјҢC1еӨ§еӨҡжҳҜеҲҶеҢ–е·®зҡ„иӮҝзҳӨпјҢиҖҢC3гҖҒC4дёәеҲҶеҢ–еҘҪзҡ„иӮҝзҳӨпјӣC2еұ…дёӯгҖӮйҡҸеҗҺпјҢTakuechiзӯүеҜ№149дҫӢNSCLC(90дҫӢи…әзҷҢ)иҝӣиЎҢ18 175дёӘеҹәеӣ иЎЁиҫҫеҲҶжһҗпјҢеҸ‘зҺ°и…әзҷҢеҸҜд»ҘеҲҶдёә2зұ»пјҢз»Ҳжң«е‘јеҗёеҚ•дҪҚеһӢ(TRU)е’Ңйқһз»Ҳжң«е‘јеҗёеҚ•дҪҚеһӢпјҢеүҚиҖ…жңүжӣҙй«ҳзҡ„EGFRзӘҒеҸҳе’Ңжӣҙе·®зҡ„йў„еҗҺгҖӮ
+ O, T1 N7 D& W& y4 ~/ Z6 W2 G( @- k S* A9 d
йҡҸеҗҺBeerдҪҝз”Ёеҫ®йҳөеҲ—жҠҖжңҜеҜ№и…әзҷҢеҲҶжһҗпјҢеҸ‘зҺ°дҪҝз”Ё50дёӘеҹәеӣ еҸҜд»Ҙе°ҶжӮЈиҖ…еҲҶдёәй«ҳеҚұйҷ©е’ҢдҪҺеҚұйҷ©2зұ»пјҢеҸҜд»ҘжҳҺжҳҫеҲӨж–ӯж—©жңҹиӮәзҷҢзҡ„з”ҹеӯҳж—¶й—ҙгҖӮPottiзӯүеҜ№198дҫӢж—©жңҹNSCLCеҲҶжһҗеҸ‘зҺ°пјҢдҪҝз”ЁеӨҡеҹәеӣ иЎЁиҫҫж ҮзӯҫвҖ”вҖ”metageneпјҢеҸҜд»Ҙйў„и®ЎжӮЈиҖ…жҳҜеҗҰеҸ‘з”ҹеӨҚеҸ‘пјҢе…·жңү72%~79%зҡ„еҮҶзЎ®жҖ§гҖӮд№ҹжңүз ”з©¶еҸ‘зҺ°пјҢдҪҝз”Ё128дёӘеҹәеӣ зҡ„иЎЁиҫҫж ҮзӯҫеҸҜд»Ҙйў„и®ЎиӮәзҷҢзҡ„жӮЈиҖ…жҳҜеҗҰеҸ‘з”ҹиҪ¬з§»гҖӮ) [2 X- ?1 j; M: |; O9 K' P- f2 N
! E2 ~. |- V# A, Z- x% d
然иҖҢпјҢеҫ®йҳөеҲ—еңЁдёҙеәҠдёҠдҪҝз”ЁеҸ—еҲ°йҷҗеҲ¶гҖӮиҝ‘жқҘпјҢChenзӯүдҪҝз”ЁRT-PCRж–№жі•еҲҶжһҗдәҶ125дҫӢNSCLCжӮЈиҖ…пјҢд»Һ672дёӘе…·жңүдҫөиўӯзү№зӮ№еҹәеӣ дёӯзӯӣжҹҘ16дёӘдёҺз”ҹеӯҳжңүе…ізҡ„еҹәеӣ пјҢеҸ‘зҺ°DUSP6гҖҒMMDгҖҒSTAT1гҖҒERBB3е’ҢLCKжҳҜ5дёӘй«ҳеҚұйҷ©еҹәеӣ пјҢжҳҜжӮЈиҖ…ж— еӨҚеҸ‘з”ҹеӯҳж—¶й—ҙзҡ„зӢ¬з«Ӣйў„еҗҺеӣ еӯҗгҖӮ
: v, x, v, |0 m% t4 B1 r z( W
, T" P! B% } j 2006е№ҙпјҢBildзӯүйҮҢзЁӢзў‘зҡ„з ”з©¶еҸ‘зҺ°пјҢе®Ңж•ҙзҡ„зҷҢеҹәеӣ йҖҡи·ҜжҝҖжҙ»зҠ¶жҖҒд№ҹеҸҜжҢҮеҜјзҷҢз—Үзҡ„еҲҶзұ»е’Ңйқ¶еҗ‘жҖ§жІ»з–—гҖӮдҪңиҖ…еҜ№NSCLCзҡ„з ”з©¶еҸ‘зҺ°пјҢRasйҖҡи·Ҝзҡ„зҠ¶жҖҒжҳҺжҳҫдёҺз»„з»ҮеӯҰдәҡеһӢзӣёе…іпјҢеӨ§еӨҡж•°и…әзҷҢж Үжң¬еҗҢйіһзҷҢзӣёжҜ”пјҢеӯҳеңЁRasзҡ„и°ғиҠӮејӮеёёгҖӮиҝӣдёҖжӯҘдҪҝз”ЁиҒҡзұ»еҲҶжһҗеҸ‘зҺ°пјҢеҸҜд»Ҙе°ҶиӮәзҷҢеҲҶдёәRasйҖҡи·ҜдҪҺжҝҖжҙ»е’ҢMycгҖҒE2F3гҖҒОІ-cateninе’ҢScrй«ҳжҝҖжҙ»зҡ„з»„пјҢRasйҖҡи·Ҝй«ҳжҝҖжҙ»е’ҢMycгҖҒE2F3гҖҒОІ-cateninе’ҢScrдҪҺжҝҖжҙ»зҡ„з»„д»ҘеҸҠRasйҖҡи·Ҝй«ҳжҝҖжҙ»е’ҢMycгҖҒE2F3гҖҒОІ-cateninе’ҢScrй«ҳжҝҖжҙ»зҡ„з»„пјҢдёҚз®Ўз»„з»Үзұ»еһӢеҰӮдҪ•пјҢйҖҡи·Ҝй«ҳеәҰжҝҖжҙ»з»„зҡ„жӮЈиҖ…жңүжӣҙе·®зҡ„йў„еҗҺ(19.7vs51.3жңҲ)гҖӮеҗҢд»ҘеүҚзҡ„еҫ®йҳөеҲ—еҲҶжһҗеӨҡдёӘиҒ”зі»дёҚжҳҺжҳҫзҡ„еҹәеӣ иҝӣиЎҢзҷҢз—ҮеҲҶзұ»дёҚеҗҢпјҢе®Ңж•ҙзҡ„зҷҢеҹәеӣ йҖҡи·ҜжҝҖжҙ»зҠ¶жҖҒеҸҜд»ҘжҸҗдҫӣж–°зҡ„жІ»з–—жңәдјҡгҖӮеҰӮдҪҝз”ЁеӨҡдёӘиҚҜзү©жҲ–еӨҡйқ¶зӮ№иҚҜзү©еҜ№е…ЁйҖҡи·Ҝзү№ејӮжҖ§жІ»з–—пјҢд№ҹи®ёеҸҜд»Ҙи§ЈеҶізӣ®еүҚйқ¶еҗ‘иҚҜзү©зҡ„иҒ”еҗҲ治疗并дёҚжҜ”еҚ•иҚҜз–—ж•ҲеҘҪзҡ„й—®йўҳгҖӮ8 X- E& M4 b/ V1 j' Y: ^6 P# M
зӣ®еүҚе·Іжңүз ”з©¶дҪҝз”ЁmiRNAиЎЁиҫҫж Үеҝ—еҲҶзұ»зҷҢз—ҮгҖҒеҲӨж–ӯзҷҢз—ҮжӮЈиҖ…зҡ„йў„еҗҺгҖӮLuзӯүиҝ‘жқҘз ”з©¶еҸ‘зҺ°пјҢеҮҶзЎ®еҲҶзұ»дәәзұ»жүҖжңүзҷҢз—Үд»…д»…йңҖиҰҒеҫҲе°‘зҡ„miRNA(зәҰ200дёӘеҹәеӣ )гҖӮжқҘжәҗдәҺдёҚеҗҢз»„з»Үзҡ„зҷҢз—ҮдҪҝз”Ёе…¶иЎЁиҫҫзҡ„miRNAеҸҜд»ҘеҪ’з»“дёәдёҖзұ»пјҢеҸҜиғҪеҜјиҮҙиӮҝзҳӨзҡ„еҲҶзұ»жҳҜдҫқжҚ®е…¶иғҡиғҺиө·жәҗиҖҢдёҚжҳҜеҷЁе®ҳгҖӮеҰӮдёҠзҡ®жқҘжәҗзҡ„зҷҢз—ҮпјҢз»“иӮ гҖҒиӮқи„ҸгҖҒиғ°и…әе’ҢиғғпјҢеҸҜд»ҘеҪ’дёәдёҖзұ»пјӣиҖҢйҖ иЎҖз»Ҷиғһиө·жәҗзҡ„иӮҝзҳӨд№ҹеҪ’дёәдёҖзұ»гҖӮ
$ j9 w1 u' ^1 M, x" ?
; }# u( e' k- }. r TakamizawaзӯүжҸҗдҫӣдәҶжӣҙзӣҙжҺҘзҡ„иҜҒжҚ®пјҢеңЁдәәзұ»иӮәзҷҢдёӯпјҢlet-7еӯҳеңЁжҳҺжҳҫдёӢи°ғпјҢиҖҢдё”let-7иЎЁиҫҫж°ҙе№ідҪҺзҡ„NSCLCзҡ„жӮЈиҖ…йў„еҗҺжӣҙе·®пјҢжүӢжңҜеҗҺз”ҹеӯҳж—¶й—ҙзҡ„жӣҙзҹӯпјҢжҸҗзӨәlet-7е…·жңүиҜҠж–ӯж„Ҹд№үгҖӮNozomuзӯүжңҖиҝ‘дҪҝз”Ёеҫ®йҳөеҲ—з ”з©¶иӮәзҷҢзҡ„miRNAзҡ„иЎЁиҫҫж Үеҝ—еҸ‘зҺ°пјҢmiRNAдёҺеҢ…жӢ¬в… жңҹеңЁеҶ…зҡ„иӮәи…әзҷҢжӮЈиҖ…зҡ„з”ҹеӯҳж—¶й—ҙеӯҳеңЁиҒ”зі»пјҢй«ҳmir-155е’ҢдҪҺlet-7a-2иЎЁиҫҫдёҺжӮЈиҖ…зҡ„йў„еҗҺе·®еӯҳеңЁиҒ”зі»гҖӮе°ҶжқҘпјҢеҜ№жқҘжәҗдәҺжҜҸдёҖзұ»иӮҝзҳӨпјҢеҸҜд»Ҙе»әз«ӢmiRNAиЎЁиҫҫж–Үеә“жҲ–miRNAиЎЁиҫҫж Үеҝ—пјҢжҢҮеҜјзҷҢз—Үзҡ„иҜҠж–ӯе’ҢжІ»з–—гҖӮз”ұдәҺmiRNAеҸҜд»Ҙз®ҖеҚ•зҡ„д»ҺзҰҸ尔马жһ—еӣәе®ҡпјҢзҹіиңЎеҢ…еҹӢзҡ„з»„з»ҮдёӯжҸҗеҸ–гҖӮ
5 P2 Y. w* a* S7 Y3 M. E7 c! X5 {1 c
дә”гҖҒ з”ҹзү©ж Үеҝ—зү©(еҹәеӣ з»„гҖҒиӣӢзҷҪз»„ж ҮзӯҫпјүдёҺNSCLCзҡ„еҢ–з–—иҚҜзү©йҖүжӢ©! W: }7 r4 e, _( n3 e! J4 `9 W
. U% @* F, z: I/ r# @$ R+ Z
еҹәеӣ з»„ж Үзӯҫ(Signature)ејҖе§Ӣз”ЁдәҺзҷҢз—Үзҡ„еҲҶзұ»пјҢзӣ®еүҚпјҢеҸҲејҖе§Ӣз”ЁдәҺжҢҮеҜјз»ҶиғһжҜ’еҢ–з–—иҚҜзү©зҡ„йҖүжӢ©гҖӮDanзӯүдҪҝз”Ё39дёӘиӮҝзҳӨз»Ҷиғһзі»еҲҶжһҗдәҶ55з§ҚеҢ–з–—иҚҜзү©еӨ„зҗҶеҗҺзҡ„еҹәеӣ иЎЁиҫҫи°ұпјҢеҸ‘зҺ°дәҶ50дёӘдёҺеҢ–з–—ж•Ҹж„ҹжҖ§зӣёе…ізҡ„еҹәеӣ пјҢжҸҗзӨәеҹәеӣ з»„ж ҮзӯҫеҸҜд»ҘдҪңдёәз”ҹзү©ж Үеҝ—зү©йў„жөӢиӮҝзҳӨеҢ–з–—иҚҜзү©зҡ„ж•Ҹж„ҹжҖ§гҖӮ8 P/ X6 m* k5 m7 C" D
7 H+ o3 ^3 M9 H. M) L7 ? 2006е№ҙPottiзӯүеҜ№еҢ…жӢ¬иӮәзҷҢз»Ҷиғһзі»зҡ„з ”з©¶еҸ‘зҺ°пјҢдҪҝз”Ёеҫ®йҳөеҲ—еҲҶжһҗж–№жі•пјҢеҗ«жңү50дёӘеҹәеӣ зҡ„иЎЁиҫҫеҸҜд»Ҙйў„и®ЎиӮәзҷҢз»Ҷиғһзі»еҜ№зҙ«жқүйҶҮзҡ„ж•Ҹж„ҹжҖ§пјҢеҜ№дәҺдёҙеәҠж Үжң¬еҮҶзЎ®зҺҮеҸҜиҫҫеҲ°80%гҖӮеҗҢж ·пјҢеҹәеӣ з»„иЎЁиҫҫж Үзӯҫд№ҹеҸҜйў„и®Ўе…¶д»–иҚҜзү©пјҢеҰӮйҳҝйңүзҙ гҖҒ5-FuгҖҒVp-16гҖҒCTXе’ҢжӢ“жҷ®жӣҝеә·пјҢиҖҢдё”иҝҳеҸҜйў„и®ЎиҒ”еҗҲеҢ–з–—зҡ„ж•Ҹж„ҹжҖ§гҖӮиҝҷдәӣеҹәеӣ еҢ…жӢ¬еҫ®з®ЎиӣӢзҷҪгҖҒTP53гҖҒдәҡз”Іеҹәеӣӣж°ўеҸ¶й…ёиҝҳеҺҹй…¶гҖҒеӨ–еҲҮдҝ®еӨҚеҹәеӣ (ERCC4)гҖҒRbйҖҡи·Ҝеҹәеӣ е’ҢBCL2зӯүгҖӮ2007е№ҙпјҢJulianзӯүдҪҝз”Ёжӣҙж–°зҡ„SiRNAж–Үеә“жҠҖжңҜеҜ»жүҫеҶіе®ҡзҙ«жқүйҶҮе’Ңе…¶д»–з»ҶиғһжҜ’иҚҜзү©ж•Ҹж„ҹжҖ§зҡ„еҹәеӣ пјҢж ·жң¬еҢ…жӢ¬NSCLCзҡ„3дёӘиӮҝзҳӨз»Ҷиғһзі»пјҢеҸ‘зҺ°дәҶеӨҡдёӘзҙ«жқүйҶҮиҖҗиҚҜзҡ„еҹәеӣ пјҢеҢ…жӢ¬еҸӮдёҺзәәй”ӨдҪ“з»„иЈ…жЈҖйӘҢзӮ№зҡ„еҹәеӣ пјҢBUB1BгҖҒAURKBпјӣд»ҘеҸҠзҘһз»Ҹй…°иғәиҪ¬иҝҗе’ҢеҗҲжҲҗзҡ„еҹәеӣ пјҢCOL4A3BPгҖҒОІ-и‘Ў(иҗ„)зі–иӢ·й…¶еҹәеӣ гҖӮ: x# t( I$ l& [- x$ v8 m9 V
* R+ `! y# b+ ?* }, B жңҖиҝ‘пјҢTaguchiзӯүдҪҝз”ЁиЎҖжё…ж Үжң¬зҡ„иҙЁи°ұеҲҶжһҗеҸ‘зҺ°дәҶйў„жөӢEGFRTKIж•Ҹж„ҹжҖ§зҡ„з”ҹзү©ж Үеҝ—гҖӮдҪңиҖ…е…ҲеҜ№139дҫӢдҪҝз”Ёgefitinibзҡ„NSCLCиҝӣиЎҢиҜ•йӘҢеҲҶжһҗпјҢеҸ‘зҺ°дәҶ8дёӘзү№ејӮзҡ„иӣӢзҷҪm/zзү№еҫҒ(з”ҹзү©ж Үеҝ—)пјҢеҲӨж–ӯйў„еҗҺзҡ„еҮҶзЎ®жҖ§иҫҫ97.1%гҖӮ然еҗҺеҜ№2дёӘзӢ¬з«Ӣзҡ„дҪҝз”ЁErlotinibжҲ–Gefitinibзҡ„дәәзҫӨиҝӣиЎҢзЎ®и®ӨпјҢеҸ‘зҺ°дҪҝз”ЁжӯӨж Үеҝ—пјҢеҘҪйў„еҗҺзҡ„жӮЈиҖ…дёӯдҪҚз”ҹеӯҳж—¶й—ҙдёә207е’Ң306еӨ©пјҢе·®йў„еҗҺзҡ„дёә92е’Ң107еӨ©гҖӮиЎҖжё…ж Үжң¬дёҙеәҠжҳ“еҫ—пјҢжүҖд»ҘжӯӨз ”з©¶е…·жңүйҮҚеӨ§ж„Ҹд№үпјҢдёәNSCLCдёӘдҪ“еҢ–жІ»з–—зҡ„зңҹжӯЈдёҙеәҠе®һи·өејҖеҗҜдәҶдёҖжүҮеӨ§й—ЁгҖӮ
! X. D" _) [3 s
, Z5 g1 {0 C( ~1 `# t. L жҖ»д№ӢпјҢе°ҶжқҘеҜ№NSCLCжІ»з–—зҡ„йҖүжӢ©пјҢд№ҹи®ёе°ҶжқҘеә”йҮҮеҸ–еҰӮдёӢжӣҙеҠ дёӘдҪ“еҢ–зҡ„жЁЎејҸ(еӣҫ1)гҖӮ- r/ E: X" _7 i
3 T3 Z; W8 q+ g1 p8 Q0 W0 \9 K& P; o! m
& r; a& ^+ \4 G/ o7 h; {* m# q
еӣҫ1 жңӘжқҘNSCLCжІ»з–—зҡ„йҖүжӢ©
4 `' y( Y% `7 f5 { }* k5 P1 J% @* U* e l
еҪ“然пјҢдёҠиҝ°з ”究д№ҹеӯҳеңЁдёҚи¶іпјҢеҰӮжңӘеҫ—еҲ°и®ҫи®ЎдёӘеҫҲеҘҪзҡ„йҡҸжңәеҜ№з…§зҡ„еүҚзһ»жҖ§з ”究иҜҒе®һпјҢиҖҢдё”еҗ„з§Қеҫ®йҳөеҲ—зҡ„еҲҶжһҗе№іеҸ°д№ҹйңҖиҰҒз»ҹдёҖгҖӮ
) ~! W3 `$ w4 t' H
) I) w5 n$ ~7 ?0 T- y6 n3 ? е…ӯгҖҒ з”ҹзү©ж Үеҝ—зү©дёҺиӮәзҷҢзҡ„дёӘдҪ“еҢ–жІ»з–—1 j6 r* | }) }% l. @
* I# o3 ^/ k7 s$ L: p4 Q (дёҖ) EGFRзӘҒеҸҳдёҺNSCLC
$ Z% k4 ^& v$ A; N2 C0 `
6 L5 E9 A s, Z+ T9 a& K иЎЁзҡ®з”ҹй•ҝеӣ еӯҗеҸ—дҪ“(epidermal growth factor receptorпјҢEGFR)е…·жңүеҸ—дҪ“й…Әж°Ёй…ёжҝҖй…¶жҙ»жҖ§(receptor tyrosinekinaseпјҢRTK)гҖӮEGFRдёәи·ЁиҶңеҸ—дҪ“пјҢз”ұз»ҶиғһеӨ–зҡ„й…ҚдҪ“з»“еҗҲеҢәеҹҹе’Ңз»ҶиғһеҶ…зҡ„й…Әж°Ёй…ёжҝҖй…¶жҙ»жҖ§еҢәеҹҹз»„жҲҗгҖӮEGFRзҡ„е°ҸеҲҶеӯҗжҠ‘еҲ¶еүӮдё»иҰҒжңү2дёӘвҖ”вҖ”Gefitinibе’ҢErlotinibпјҢеҲҶеҲ«дәҺ2003е’Ң2004е№ҙеңЁзҫҺеӣҪиҺ·FDAжү№еҮҶдёҠеёӮпјҢз”ЁдәҺеҜ№дј з»ҹеҢ–з–—ж— ж•Ҳзҡ„жҷҡжңҹNSCLCжӮЈиҖ…гҖӮ0 \# Q8 I+ S/ h+ i: n% D
3 N. }% n9 l: c d# z. K9 S
GefitinibгҖҒErlotinibзҡ„й«ҳж•Ҹж„ҹжҖ§дёҺEGFRзҡ„дҪ“з»ҶиғһжҖ§(somaticпјҢдёҚеҗҢдәҺе…ҲеӨ©жҖ§)зӘҒеҸҳжҳҺжҳҫзӣёе…ігҖӮеңЁйқһйҖүжӢ©жҖ§NSCLCж Үжң¬пјҢEGFRзӘҒеҸҳзҺҮеңЁеҢ—зҫҺе’ҢиҘҝ欧дёә10%пјҢиҖҢеңЁдёңдәҡдёә30%~50%гҖӮEGFRзӘҒеҸҳеқҮеҸ‘з”ҹдәҺ4дёӘеӨ–жҳҫеӯҗдёӯ(18-21)пјҢзј–з ҒEGFRйғЁеҲҶзҡ„й…Әж°Ёй…ёжҝҖй…¶еҢәеҹҹ(е…ЁйғЁз”ұ18-24зј–з Ғ)гҖӮдё»иҰҒзӘҒеҸҳжңүпјҡеӨ–жҳҫеӯҗ19зҡ„зјәеӨұ(4дёӘж°Ёеҹәй…ёпјҡLREA)пјҢеӨ–жҳҫеӯҗ21зҡ„L858RзӮ№зӘҒеҸҳпјҢеӨ–жҳҫеӯҗ18зҡ„G719SзӮ№зӘҒеҸҳпјҢд»ҘеҸҠеӨ–жҳҫеӯҗ20зҡ„770жҸ’е…ҘзӘҒеҸҳ(е…·дҪ“еҰӮеӣҫ2)гҖӮ
6 Q- V3 f4 x% k
# k0 F- z9 }! N/ N' I3 M1. EGFRзӘҒеҸҳдёҺNSCLCжІ»з–—зҡ„й«ҳж•Ҹж„ҹжҖ§
) F3 \# w1 L* t a- b6 v; I4 r, f
4 \9 ]( Y! G0 t: V EGFRжҝҖй…¶еҢәеҹҹзҡ„зӘҒеҸҳд»…йҷҗдәҺжҹҗдәӣдәҡеһӢзҡ„NSCLCпјҢеҰӮдёҚеҗёзғҹзҡ„дәҡжҙІеҘіжҖ§пјҢз»„з»Үзұ»еһӢдёәи…әзҷҢе’Ңж”Ҝж°”з®ЎиӮәжіЎзҷҢгҖӮжңүзӘҒеҸҳзҡ„жӮЈиҖ…еҜ№EGFR-TKIжІ»з–—еҸҚеә”жӣҙеҘҪгҖӮ(еӣҫ2). H9 X+ s9 c9 g* I! h
" A! Z% e. `) E. g: `% c1 q/ Q Q еӨ§еӨҡж•°еӣһйЎҫжҖ§з ”究已з»ҸеҸ‘зҺ°пјҢEGFRзӘҒеҸҳзҡ„NSCLCдёӯпјҢ50%~80%еҜ№GefitinibгҖҒErlotinibжңүеҸҚеә”пјҢиҖҢдё”иҝ‘жқҘжқҘиҮӘдәҡжҙІзҡ„жҠҘйҒ“жӣҙй«ҳпјҢи¶…иҝҮ75%пјҢиҖҢ GefitinibгҖҒErlotinibдёҚж•Ҹж„ҹзҡ„жӮЈиҖ…дёӯзӘҒеҸҳзҺҮдёә7%гҖӮжҸҗзӨәзӘҒеҸҳжҳҜGefitinibгҖҒErlotinibж•Ҹж„ҹжҖ§еўһй«ҳзҡ„еҺҹеӣ (е…·дҪ“и§Ғеӣҫ2)гҖӮиҷҪ然еӨ§еӨҡж•°EGFRзӘҒеҸҳиў«и®ӨдёәжҳҜTKIжҲҸеү§жҖ§дёҙеәҠз–—ж•Ҳзҡ„еҹәзЎҖпјҢдҪҶзәҰ10%~20%дёҚеӯҳеңЁEGFRзӘҒеҸҳзҡ„жӮЈиҖ…еҜ№GefitinibжңүйғЁеҲҶеҸҚеә”пјҢжҸҗзӨәEGFRзӘҒеҸҳеҜ№TKIз–—ж•ҲдёҚжҳҜдёҖдёӘеҝ…дёҚеҸҜе°‘зҡ„еҶіе®ҡеӣ зҙ гҖӮе…¶д»–зҡ„еҲҶеӯҗејӮеёёпјҢеҰӮEGFRеҹәеӣ жү©еўһ(еҹәеӣ жӢ·иҙқж•°еўһеҠ )дёҺTKIзҡ„й«ҳж•Ҹж„ҹжҖ§зӣёе…іпјҢеҰӮHirshзӯүжҠҘйҒ“пјҢй«ҳEGFRжӢ·иҙқж•°зҡ„жӮЈиҖ…жңүжӣҙеҘҪзҡ„йў„еҗҺгҖӮ5 y* D4 E& z0 o6 C7 v+ P' e8 w7 _
иҝӣдёҖжӯҘз ”з©¶иҝҳеҸ‘зҺ°пјҢдёҚеҗҢзҡ„зӘҒеҸҳдёҺжӮЈиҖ…зҡ„дёҙеәҠжІ»з–—ж•Ҳжһңй«ҳдҪҺеӯҳеңЁдёҖе®ҡзҡ„иҒ”зі»пјҢеҰӮеӨ–жҳҫеӯҗ21зӮ№зӘҒеҸҳ(L585R)дёҺеӨ–жҳҫеӯҗ19зјәеӨұжҖ§зӘҒеҸҳзӣёжҜ”пјҢеҗҺиҖ…еҸҜиғҪеҜ№GefitinibгҖҒErlotinibжңүжӣҙеҘҪеҸҚеә”гҖӮ
( O' I; W0 X. n# E0 G: {
9 _, w- g. d S, a/ E: i0 B/ F% l4 O E' q* E* L: O
' j5 F- P O" @; @# f" Y: c 2. EGFRзӘҒеҸҳж”№еҸҳGefitinibгҖҒErlotinibж•Ҹж„ҹжҖ§зҡ„еҺҹеӣ EGFRзӘҒеҸҳ并没жңүеҪұе“ҚдёҺGefitinibгҖҒErlotinibз»“еҗҲзҡ„иғҪеҠӣгҖӮе…¶ж”№еҸҳиҚҜзү©ж•Ҹж„ҹжҖ§зҡ„еҺҹеӣ еҸҜд»ҘйҖҡиҝҮзҷҢеҹәеӣ дҫқиө–(oncogene addiction)жЁЎеһӢжқҘи§ЈйҮҠгҖӮ: \! ~& u/ p, ?: B! `
0 A( B$ A% W' W( Y йҖҡиҝҮRas-Raf-MEK-ERK1/ERK2пјҢPI3K-AktпјҢSTAT3/STAT5йҖҡи·ҜпјҢEGFRзӘҒеҸҳй«ҳеәҰжҝҖжҙ»EGFRи°ғиҠӮзҡ„жҠ—еҮӢдәЎе’Ңеўһж®–дҝЎеҸ·пјҢеҜјиҮҙзҷҢз—Үз»ҶиғһеҸҳеҫ—дҫқиө–жӯӨдҝЎеҸ·д»Ҙз»ҙжҢҒе…¶з”ҹеӯҳвҖ”вҖ”еҚіз»Ҷиғһе…·жңүзҷҢеҹәеӣ (зӘҒеҸҳзҡ„EGFR)дҫқиө–зҡ„зү№еҫҒпјӣеҪ“дҪҝз”Ёзү№ејӮжҖ§TKIйҳ»ж–ӯEGFRдҝЎеҸ·еҗҺпјҢе°Ҷж¶ҲйҷӨе…¶еўһж®–жҖ§еҪұе“Қе’Ңиҫ“еҮәз”ҹеӯҳдҝЎеҸ·пјҢеҜјиҮҙиӮҝзҳӨз»Ҷиғһжӯ»дәЎгҖӮзӣёеҸҚпјҢжӯЈеёёз»ҶиғһжҲ–йқһEGFRдҫқиө–зҡ„иӮҝзҳӨз»Ҷиғһ(еҜ№GefitinibгҖҒErlotinibж— еҸҚеә”)дёҚеҸ—еҪұе“ҚпјҢеӣ дёәз”ҹеӯҳиҝҳеҸ—е…¶д»–еҹәеӣ й©ұдҪҝпјҢжҲ–иҖ…еңЁEGFRжҠ‘еҲ¶еҗҺиғҪиў«е…¶д»–зҡ„еҹәеӣ йҖҡи·ҜжүҖејҘиЎҘгҖӮ
: z" s% h2 _+ K0 j2 U/ R6 P& o+ T) a, @
еңЁзҷҢеҹәеӣ дҫқиө–з»ҶиғһпјҢе…ій”®зҷҢеҹәеӣ зҡ„жҖҘжҖ§еӨұжҙ»(еҰӮTKIжҠ‘еҲ¶EGFR)еҜјиҮҙеҮәзҺ°зҷҢеҹәеӣ дј‘е…Ӣ(oncogene shock)пјҢеҚіеҮӢдәЎдҝЎеҸ·зҡ„иҫ“еҮәеӨ§дәҺз”ҹеӯҳдҝЎеҸ·пјҢдҝғдҪҝеҮӢдәЎеҸ‘з”ҹгҖӮзҷҢеҹәеӣ дј‘е…ӢеҸҜд»Ҙи§ЈйҮҠпјҡз»ҶиғһжҜ’еҢ–з–—жҚҹдјӨз»ҶиғһDNAпјҢеҜјиҮҙз»Ҷиғһе‘ЁжңҹеҒңжӯўвҖ”вҖ”еҮӢдәЎе’Ңз”ҹеӯҳдҝЎеҸ·зҡ„иҫ“еҮәеқҮеҮҸејұпјҢз»“жһңз»ҶиғһдёҚеҶҚдҫқиө–жҹҗдёӘзҷҢеҹәеӣ пјҢеӣ жӯӨдёҺTKIиҒ”еҗҲжІ»з–—ж—¶зҡ„з–—ж•Ҳ并дёҚеӯҳеңЁеҚҸеҗҢгҖӮ& Y5 p; G! r) \+ ^. n/ ~ x2 w, f
/ j; R* n% w% `4 a$ S
еҜ№дәҺзӘҒеҸҳеҜјиҮҙGefitinibгҖҒErlotinibж•Ҹж„ҹжҖ§еўһй«ҳпјҢзҷҢеҹәеӣ дҫқиө–д№ҹ并дёҚе®Ңе…ЁеҗҲзҗҶи§ЈйҮҠпјҢеӣ дёәEGFRеҚ•е…ӢйҡҶжҠ—дҪ“cetuximabиҷҪ然еҸҜд»ҘжҠ‘еҲ¶EGFRеҜјиҮҙеҮӢдәЎе’Ңз”ҹеӯҳдҝЎеҸ·иҫ“еҮәдёӢйҷҚпјҢдҪҶе…¶з–—ж•ҲдёҺзӘҒеҸҳж— е…ігҖӮиҖҢдё”жңҖиҝ‘еҸ‘зҺ°зӘҒеҸҳеҗҺзҡ„EGFRеҜ№GefitinibдәІеҗҲеҠӣжҳҺжҳҫжҸҗй«ҳпјӣжҸҗзӨәNSCLCз»ҶиғһеӯҳеңЁе…¶д»–зҡ„еӣ зҙ еҸӮдёҺз–—ж•Ҳе·®ејӮзҡ„еҪўжҲҗгҖӮ
# w' }+ L. ]) G/ T- P- f- V9 F( r: J3 e2 E% m X
3.EGFRзӘҒеҸҳдёҺNSCLCжІ»з–—зҡ„иҖҗиҚҜ: h& C: d4 k! Q6 T/ E, Z5 ]
6 i5 b1 {/ @- \, Q
EGFRзӘҒеҸҳ并дёҚйғҪжҳҜеҜјиҮҙGefitinibгҖҒErlotinibзҡ„ж•Ҹж„ҹжҖ§жҸҗй«ҳпјҢиҝҳжҳҜEGFR-TKIиҖҗиҚҜзҡ„еҺҹеӣ д№ӢдёҖгҖӮ W, [$ w4 l3 W4 J8 r3 i- c8 G7 A
- H) S) S1 d0 C& L- Q2 M (1) еҺҹеҸ‘иҖҗиҚҜ+ l0 o; s& ^/ D; M* }) f; v
' d. {: X1 q& F0 }' N$ f- ^ иҝ‘жқҘзҡ„з ”з©¶жҸҗзӨәпјҢEGFRеӨ–жҳҫеӯҗ20зҡ„жҸ’е…ҘжҖ§зӘҒеҸҳ(D770_N771)еҸҜд»ҘдҪҝеҸ—дҪ“еҜ№EGFR-TKIзҡ„ж•Ҹж„ҹжҖ§йҷҚдҪҺ100еҖҚпјҢдёҙеәҠдёҠд№ҹеҸ‘зҺ°е…·жңүжӯӨзӘҒеҸҳзҡ„жӮЈиҖ…еҜ№EGFR-TKIжІ»з–—еҸҚеә”дёҚжҳҺжҳҫгҖӮT790MзӘҒеҸҳеҫҲе°‘и§ҒпјҢеӨ§еӨҡж•°еҜјиҮҙиҺ·еҫ—жҖ§иҖҗиҚҜпјҢдҪҶд№ҹдёҺеҺҹеҸ‘иҖҗиҚҜзӣёе…іпјӣиҖҢдё”T790MзӘҒеҸҳдёҺе…¶д»–ж•Ҹж„ҹжҖ§зӘҒеҸҳпјҢеҰӮL585RеҗҢж—¶еӯҳеңЁж—¶пјҢзҷҢз—Үз»ҶиғһеҜ№EGFR-TKIд№ҹжҳҜжҠөжҠ—зҡ„гҖӮеҜ№дәҺеӨ§еӨҡж•°еҜ№EGFR-TKIиҖҗиҚҜзҡ„NSCLCиҖҢиЁҖпјҢе…¶иӮҝзҳӨзҡ„еҪўжҲҗеҫҲеҸҜиғҪдёҚжҳҜдҫқиө–EGFRеҜјиҮҙгҖӮ1 N) s4 O% p+ U
# \9 z. Y" A. }) V5 y KRASжҳҜEGFRдёӢжёёзҡ„е…ій”®и°ғиҠӮеҲҶеӯҗпјҢеӨ§зәҰ15%~30%зҡ„NSCLCеӯҳеңЁKRASеҜҶз Ғеӯҗ12е’Ң13зӘҒеҸҳпјҢдёҺжӮЈиҖ…зҡ„йў„еҗҺе·®зӣёе…ігҖӮжңүз ”з©¶жҸҗзӨәKRASзӘҒеҸҳеҸҜиғҪжҳҜGefitinibгҖҒErlotinibеҺҹеҸ‘иҖҗиҚҜзҡ„еҺҹеӣ гҖӮдёҖиҲ¬жқҘи®ІпјҢKRASе’ҢEGFRзӘҒеҸҳNSCLCжҳҜзӣёдә’жҺ’ж–Ҙзҡ„пјҢEGFRзӘҒеҸҳдё»иҰҒи§ҒдәҺдёҚеҗёзғҹиҖ…пјҢиҖҢKRASзӘҒеҸҳжӣҙеёёи§ҒдәҺеҗёзғҹзӣёе…ізҡ„зҷҢз—ҮгҖӮеӣ дёәKRASзӘҒеҸҳжҖ»жҳҜеҸ‘з”ҹдәҺе…·жңүйҮҺз”ҹеһӢEGFRзҡ„NSCLCдёӯпјҢжүҖд»Ҙйҡҫд»ҘеҢәеҲҶеҜ№EGFR-TKIдёҚж•Ҹж„ҹеҲ°еә•жҳҜеӣ дёәKRASзӘҒеҸҳпјҢиҝҳжҳҜеӣ дёәж— EGFRзӘҒеҸҳгҖӮ" K# Y/ I$ U# c) p, \- b
" a/ t7 u- Y% C. a9 \" ]' _: v: [
е…¶д»–EGFRдёӢжёёеҲҶеӯҗд№ҹдёҺGefitinibгҖҒErlotinibзҡ„иҖҗиҚҜжңүе…ігҖӮеҰӮAktзҡ„жҝҖжҙ»жҳҜиҚҜзү©дёҚж•Ҹж„ҹзҡ„дёҖдёӘж Үеҝ—гҖӮAktзҡ„жҝҖжҙ»з”ұPTENеҹәеӣ й—ҙжҺҘи°ғиҠӮпјҢNSCLCдёӯпјҢPTENдҪҺиЎЁиҫҫ(з”ұдәҺеҗҜеҠЁеӯҗз”ІеҹәеҢ–)дёҺGefitinibгҖҒErlotinibзҡ„иҖҗиҚҜзӣёе…ігҖӮIGFR1гҖҒERBB3е’ҢERBB2д№ҹеҸӮдёҺGefitinibзҡ„иҖҗиҚҜгҖӮ然иҖҢпјҢиҝ‘жқҘеӨ§ж ·жң¬з ”究еҸ‘зҺ°пјҢPTENе’ҢIGFR1зҡ„зҠ¶жҖҒдёҺGefitinibзҡ„жІ»з–—еҸҚеә”дёҚзӣёе…іпјҢжҸҗзӨәдёҠиҝ°еҲҶеӯҗеңЁиҖҗиҚҜдёӯдҪңз”Ёиҝҳеҫ…иҝӣдёҖжӯҘз ”з©¶гҖӮ
A) h3 r ]2 Q5 S
* [, X D. N1 L" @4 ] (2) иҺ·еҫ—жҖ§иҖҗиҚҜ" U8 q9 T/ l' b7 T# p4 J
3 G, N. a3 ]0 y( T е°Ҫз®ЎеңЁEGFRзӘҒеҸҳзҡ„NSCLCдёӯпјҢGefitinibгҖҒErlotinibеҸҜд»ҘиҫҫеҲ°жҲҸеү§жҖ§зҡ„ж•ҲжһңпјҢдҪҶејҖе§ӢжІ»з–—еҗҺ6~12дёӘжңҲеҸ‘з”ҹзҡ„иҖҗиҚҜжҳҺжҳҫйҷҗеҲ¶дәҶжӮЈиҖ…з”ҹеӯҳж—¶й—ҙзҡ„延й•ҝгҖӮеҜ№иҖҗиҚҜеҲҶеӯҗе’Ңз»ҶиғһжңәеҲ¶ж·ұе…ҘдәҶи§ЈжҳҜе°ҶжқҘе…ӢжңҚиҖҗиҚҜзҡ„е…ій”®гҖӮеңЁејҖе§ӢеҜ№GefitinibгҖҒErlotinibжңүеҸҚеә”дҪҶеҗҺжқҘеӨҚеҸ‘зҡ„жӮЈиҖ…(е…·жңүEGFRзӘҒеҸҳ)дёӯпјҢ еӯҳеңЁEGFRеӨ–жҳҫеӯҗ20зҡ„继еҸ‘жҖ§зӘҒеҸҳвҖ”вҖ”T790MпјҢиҖҗиҚҜзҡ„еҺҹеӣ жҳҜзӘҒеҸҳеҜјиҮҙEGFRз»“жһ„еҸ‘з”ҹеҸҳеҢ–пјҢдҪҝTKIдёҺе…¶з»“еҗҲеҮәзҺ°дҪҚйҳ»ж•Ҳеә”гҖӮеңЁ50%зҡ„TKIжІ»з–—еҗҺеӨҚеҸ‘зҡ„NSCLCдёӯеҸҜд»ҘеҸ‘зҺ°T790MзӘҒеҸҳгҖӮдёҖдәӣз ”з©¶иҖ…д№ҹи®ӨдёәпјҢиҚҜзү©жІ»з–—д№ӢеүҚT790MзӘҒеҸҳз»ҶиғһеҚіеңЁжӮЈиҖ…дҪ“еҶ…еӯҳеңЁпјҢеҪ“з»ҶиғһжҡҙйңІдәҺTKIеҗҺпјҢз”ұдәҺиҖҗиҚҜиҖҢеҜјиҮҙеҸ‘жҢҘйҖүжӢ©дҪңз”ЁпјҢдҪҝT790MзӘҒеҸҳз»Ҷиғһж•°зӣ®жҳҺжҳҫеўһеӨҡгҖӮеӣ жӯӨд№ҹиҜҙжҳҺеҺҹеҸ‘иҖҗиҚҜе’Ң继еҸ‘иҖҗиҚҜзҡ„еҢәеҲ«еҸҜиғҪд»…жҳҜT790MзӘҒеҸҳз»Ҷиғһзҡ„еӨҡе°‘иҖҢе·ІгҖӮ
# b/ o$ y4 Q0 |2 |& I
8 U, D8 e6 l5 S- Y иҺ·еҫ—жҖ§иҖҗиҚҜе…¶д»–еҸҜиғҪзҡ„жңәеҲ¶еҢ…жӢ¬пјҢEGFRиҝҗиҫ“зҡ„ж”№еҸҳпјҢзӘҒеҸҳEGFRзҡ„жү©еўһпјҢжҲ–иҖ…EGFRзҡ„дёӢжёёдҝЎеҸ·еҲҶеӯҗзҡ„й«ҳиЎЁиҫҫпјҢжҲ–иҖ…з»Ҷиғһе°ҶиҚҜзү©зҡ„з”ҹзү©жҙ»жҖ§ж”№еҸҳдәҶгҖӮз ”з©¶е·Із»ҸжҸӯзӨәпјҢеӨҡиҚҜиҖҗиҚҜиӣӢзҷҪABCG2еҸҜд»Ҙе°ҶGefitinibжіөеҮәз»ҶиғһеӨ–пјҢеӣ жӯӨеҜјиҮҙиҖҗиҚҜпјӣдҪҶд№ҹжңүз ”з©¶жҸҗзӨәпјҢGefitinibжң¬иә«еҸҜд»ҘдҪҝABCG2е’ҢABCиҪ¬иҝҗPзі–иӣӢзҷҪеӨұжҙ»гҖӮ) H3 L3 Y) Z- t$ j, x$ \% S0 c
( u8 M& x/ A# V: E% s
(дәҢ) ERCC1е’ҢRRM1дёҺNSCLCзҡ„дёӘдҪ“еҢ–жІ»з–—
' N1 x2 n* D0 {; P' |" S% ?/ K5 G; q5 z" x) Z" C( L/ k
10еӨҡе№ҙеүҚеҚіе·ІзҹҘйҒ“пјҢж ёй…ёеӨ–еҲҮдҝ®еӨҚеҸӮдёҺеҮ з§ҚзҷҢз—Үзҡ„иҖҗиҚҜпјҢеҰӮй“Ӯзұ»иҚҜзү©гҖӮERCC1жҳҜй«ҳеәҰдҝқе®Ҳзҡ„иӣӢзҷҪпјҢз»ҙжҢҒз”ҹе‘Ҫжҙ»еҠЁжүҖеҝ…йңҖгҖӮиҷҪ然е®ғжҳҜеҮ дёӘж ёй…ёеӨ–еҲҮдҝ®еӨҚиӣӢзҷҪдёӯзҡ„дёҖдёӘпјҢдҪҶжҳҜе…·жңүйҷҗйҖҹй…¶зҡ„дҪңз”ЁгҖӮ2006е№ҙпјҢOlaussenзӯүжҠҘйҒ“пјҢеңЁжүӢжңҜеҗҺдҪҝз”Ёй“Ӯзұ»иҚҜзү©дёәеҹәзЎҖзҡ„иҫ…еҠ©еҢ–з–—NSCLCдёӯпјҢзјәд№ҸERCC1иӣӢзҷҪзҡ„жӮЈиҖ…жңүжӣҙеҘҪзҡ„йў„еҗҺгҖӮERCC1йҳҙжҖ§зҡ„жӮЈиҖ…пјҢжҺҘеҸ—иҫ…еҠ©еҢ–з–—иҖ…зҡ„дёӯдҪҚз”ҹеӯҳжңҹжҜ”жҺҘеҸ—еҚ•зәҜжүӢжңҜиҖ…еўһеҠ дәҶ14дёӘжңҲ(56дёӘжңҲеҜ№42дёӘжңҲпјҢP=0.002)гҖӮиҖҢERCC1йҳіжҖ§иҖ…жҳҜеҗҰжҺҘеҸ—иҫ…еҠ©еҢ–з–—дёҺз”ҹеӯҳжңҹж— е…і(50дёӘжңҲеҜ№55дёӘжңҲпјҢP=0.40)пјӣеңЁжҺҘеҸ—еҚ•зәҜжүӢжңҜзҡ„жӮЈиҖ…дёӯпјҢERCC1йҳіжҖ§иҖ…з”ҹеӯҳжңҹиҫғERCC1йҳҙжҖ§иҖ…й•ҝгҖӮдёҠиҝ°з»“жһңиЎЁжҳҺпјҢERCC1йҳҙжҖ§зҡ„жӮЈиҖ…жӣҙиғҪд»Һеҗ«й“Ӯж–№жЎҲзҡ„иҫ…еҠ©еҢ–з–—дёӯиҺ·зӣҠгҖӮеңЁDNAеҗҲжҲҗе’Ңдҝ®еӨҚдёӯзҡ„еҸҰдёҖдёӘе…ій”®й…¶жҳҜж ёз”ҳй…ёиҝҳеҺҹй…¶M1(RRM1)пјҢж—©жңҹзҡ„з ”з©¶е·Із»ҸеҸ‘зҺ°пјҢеңЁиӮәзҷҢдёӯпјҢRRM1зҡ„иҝҮиЎЁиҫҫдёҺй“Ӯзұ»иҚҜзү©е’ҢеҗүиҘҝд»–ж»ЁиҖҗиҚҜжңүе…іпјҢ2006е№ҙпјҢCeppiзӯүеҸ‘зҺ°пјҢERCC1е’ҢRRM1зҡ„mRNAж°ҙе№ідёҺжӮЈиҖ…зҡ„йў„еҗҺе·®жҳҺжҳҫзӣёе…іпјҢдҪҝз”ЁGPж–№жЎҲеҢ–з–—гҖӮ2007е№ҙ2жңҲпјҢдҪӣе·һеқҰдјҜLee MoffittзҷҢз—Үдёӯеҝғзҡ„з ”з©¶жҢҮеҮәпјҢжҺҘеҸ—еҸҜиғҪжІ»зҷ’жҖ§жүӢжңҜзҡ„ж—©жңҹ(в… жңҹ)йқһе°Ҹз»ҶиғһиӮәзҷҢ(NSCLC)з—…жӮЈдёӯпјҢRRM1е’ҢERCC1еҗҢж—¶й«ҳиЎЁиҫҫзҡ„жӮЈиҖ…(еӨ§зәҰ30%пјҢ55/184)йў„еҗҺиүҜеҘҪпјҢеӣ жӯӨжҳҫ然дёҺд»ҘеүҚзҡ„жҠҘйҒ“зӣёеҸҚгҖӮ
/ b0 h$ q3 `$ K$ H* T8 P& }6 |3 s5 B
дҪҶжҳҜпјҢд»”з»ҶеҲҶжһҗRRM1е’ҢERCC1зҡ„еҠҹиғҪе°ұеҸҜд»ҘеҗҲзҗҶи§ЈйҮҠпјҢеҚіRRM1е’ҢERCC1еңЁдёҚеҗҢзҡ„йҳ¶ж®өе…·жңүдёҚеҗҢзҡ„и§’иүІгҖӮйҖҡиҝҮйў„йҳІзӘҒеҸҳпјҢRRM1е’ҢERCC1еҸҜеҮҸе°‘зҷҢз—Үзҡ„еҸ‘з”ҹпјҢжҲ–иҖ…йҳ»жӯўе·Із»ҸеӯҳеңЁзҡ„иӮҝзҳӨиҝӣеұ•гҖӮеӣ жӯӨеҜ№дәҺжңӘиҝӣиЎҢеҢ–з–—зҡ„ж—©жңҹNSCLCжӮЈиҖ…пјҢRRM1е’ҢERCC1жҳҜйў„еҗҺеҘҪзҡ„ж Үеҝ—гҖӮиҖҢеҜ№дәҺжҷҡжңҹNSCLCпјҢиӢҘиҝӣиЎҢеҢ–з–—еҲҷеҸҜиғҪеҜјиҮҙиҖҗиҚҜгҖӮеӣ жӯӨпјҢRRM1е’ҢERCC1еҜ№дәҺNSCLCжӮЈиҖ…жІ»з–—зҡ„йҖүжӢ©еӯҳеңЁ2з§Қжғ…еҶөпјҡж—©жңҹжӮЈиҖ…пјҢRRM1е’ҢERCC1й«ҳиЎЁиҫҫеҲҷеҸҜд»ҘдёҚиҝӣиЎҢжІ»з–—пјӣеҜ№дәҺжҷҡжңҹжӮЈиҖ…пјҢRRM1е’ҢERCC1й«ҳиЎЁиҫҫпјҢеҲҷдёҚйҖӮеҗҲдј з»ҹзҡ„еҢ–з–—пјҢеҸҜдҪҝз”ЁEGFRжҠ‘еҲ¶еүӮгҖӮ
/ j, i3 P: x1 _2 @: \4 b
! o& i# ]7 ]' {2 {* M) Y ReimanзӯүйҮҮз”Ёе…Қз–«з»„еҢ–жҠҖжңҜжЈҖжөӢJBR10з ”з©¶дёӯ265дҫӢNSCLCОІеҫ®з®ЎиӣӢзҷҪIIIзҡ„иЎЁиҫҫпјҢеҸ‘зҺ°еңЁиҫ…еҠ©еҢ–з–—з»„пјҢОІеҫ®з®ЎиӣӢзҷҪIIIй«ҳиЎЁиҫҫиҖ…жңүжӣҙй•ҝзҡ„ж— еӨҚеҸ‘з”ҹеӯҳжңҹ(P=0.002)пјҢжҸҗзӨәй•ҝжҳҘз‘һж»Ё+йЎәй“Ӯж–№жЎҲиҫ…еҠ©еҢ–з–—еҸҜиғҪдҪҝй«ҳиЎЁиҫҫОІеҫ®з®ЎиӣӢзҷҪIIIзҡ„NSCLCжӮЈиҖ…иҺ·зӣҠжӣҙеӨҡгҖӮ9 w; N* q% B2 l. t; h
6 `) R0 R L: e4 {4 ?
(дёү)EMP-1дёҺNSCLCзҡ„дёӘдҪ“еҢ–жІ»з–—
6 V4 ]( ?' q6 `/ T
, L2 S; y2 U& u' H8 y, S' q! R AnjaliзӯүеҲҶжһҗ39дҫӢдҪҝз”ЁGefitinibеҚ•иҚҜжІ»з–—зҡ„жҷҡжңҹNSCLCжӮЈиҖ…пјҢеҸ‘зҺ°дҪҺEMP-1иЎЁиҫҫзҡ„жӮЈиҖ…еҸҚеә”зҡ„еҸҜиғҪжҖ§й«ҳ(70%)пјҢиӢҘEMP-1иЎЁиҫҫеўһеҠ пјҢеҲҷеҸҚеә”зҡ„еҸҜиғҪжҖ§еҫҲдҪҺ(5%)гҖӮдёҙеәҠеҜ№GefitinibеӯҳеңЁеҸҚеә”зҡ„жӮЈиҖ…еҹәжң¬дёҚдјҡиЎЁиҫҫEMP-1пјҢиҖҢ14дёӘиЎЁиҫҫEMP-1зҡ„жӮЈиҖ…дёӯпјҢйҷӨдёҖдёӘз—…жғ…зЁіе®ҡеӨ–пјҢеқҮеҮәзҺ°з–ҫз—…иҝӣеұ•гҖӮ
# j9 ?) b) ^5 l7 O4 r' f8 }
& t0 l4 j& X( X n еҲҶжһҗ103дҫӢйқһйҖүжӢ©зҡ„иӮәзҷҢжӮЈиҖ…зҡ„ж Үжң¬пјҢеҸ‘зҺ°йіһзҷҢзҡ„EMP-1иЎЁиҫҫзҺҮдёә66%пјҢи…әзҷҢдёә40.9%гҖӮеҲҶжһҗEMP-1иЎЁиҫҫдёҺEGFRзӘҒеҸҳд№Ӣй—ҙзҡ„е…ізі»еҸ‘зҺ°пјҢиЎЁиҫҫEMP-1зҡ„жӮЈиҖ…дёҚеӯҳеңЁEGFRзӘҒеҸҳгҖӮ4 T7 C1 Q) S* \9 z- j: U5 R# P
" q/ j6 N0 o0 n C# t& Z2 m& Z
еҲҶжһҗGefitinibиҺ·еҫ—иҖҗиҚҜзҡ„жӮЈиҖ…еҸ‘зҺ°пјҢиҝҷдәӣжӮЈиҖ…дёәеңЁиҜҠж–ӯж—¶дёәBCAпјҢејҖе§ӢеҜ№GefitinibеӯҳеңЁеҸҚеә”пјҢз”ұдәҺеӯҳеңЁL858RзӘҒеҸҳпјҢGefitinibжІ»з–—еүҚзҡ„ж Үжң¬EMP-1иЎЁиҫҫеҫҲдҪҺ(е°ҸдәҺ10%)пјҢиҝҷдәӣжӮЈиҖ…жІ»з–—160еӨ©еҗҺеҮәзҺ°иҖҗиҚҜпјҢеҲҶжһҗж Үжң¬еҸ‘зҺ°пјҢжӮЈиҖ…еҮәзҺ°дәҶж–°зҡ„T790MзӘҒеҸҳпјҢеҗҢж—¶пјҢEMP-1иЎЁиҫҫзҡ„иЎЁиҫҫд№ҹжҳҺжҳҫдёҠеҚҮгҖӮжҸҗзӨәEMP-1жҳҜGefitinibеҺҹеҸ‘е’ҢиҺ·еҫ—иҖҗиҚҜзҡ„з”ҹзү©ж Үеҝ—пјҢиҖҢдёҺEGFRзҡ„жҝҖй…¶еҢәеҹҹзӘҒеҸҳж— е…ігҖӮдёҚжҳҜжүҖжңүзҡ„еҜ№Gefitinibж— еҸҚеә”зҡ„жӮЈиҖ…еӯҳеңЁEMP-1иЎЁиҫҫпјҢжҸҗзӨәGefitinibзҡ„иҖҗиҚҜеӯҳеңЁе…¶д»–жңәеҲ¶гҖӮ p$ c; F8 }5 U0 c5 R
) I* W4 g$ K8 v, b' u$ a5 i
(еӣӣ) EGFRзӘҒеҸҳдёҺNSCLCзҡ„ж”ҫз–—ж•Ҹж„ҹжҖ§4 e% U. O' w& G& a5 I$ i! r
% ?+ U1 j& n% I5 h/ ?8 V йҷӨдәҶдј—жүҖе‘ЁзҹҘзҡ„SCLCе’ҢNSCLCд№Ӣй—ҙеӯҳеңЁжҳҺжҳҫзҡ„ж”ҫз–—ж•Ҹж„ҹжҖ§е·®ејӮд№ӢеӨ–пјҢеңЁNSCLCдёӯзӣёеҗҢзҡ„з»„з»Үзұ»еһӢд№Ӣй—ҙд№ҹеӯҳеңЁжҳҺжҳҫзҡ„е·®ејӮгҖӮиӮҝзҳӨж”ҫз–—дёӘдҪ“еҢ–зҡ„е…ій”®жҳҜиғҪйў„жөӢиӮҝзҳӨеҶ…еңЁж”ҫе°„ж•Ҹж„ҹжҖ§гҖӮ
1 J6 a ~- `, K" P: R: [4 c2 d/ n ^2 M
еңЁеӨҡз§ҚиӮҝзҳӨ(еҢ…жӢ¬NSCLC)дёӯпјҢEGFRзҡ„жҝҖжҙ»е’ҢиЎЁиҫҫжҳҜж”ҫз–—ж•Ҹж„ҹжҖ§зҡ„еҶіе®ҡеӣ зҙ пјҢзӣёеҸҚпјҢзӘҒеҸҳеҠҹиғҪзҡ„дёўеӨұжҲ–дҪҝз”ЁжҠ—EGFRжІ»з–—еҸҜд»ҘеўһеҠ ж”ҫз–—зҡ„ж•Ҹж„ҹжҖ§гҖӮAmitзӯүдҪҝз”Ё19дёӘNSCLCз»Ҷиғһзі»пјҢ10дёӘе…·жңүж— зӘҒеҸҳзҡ„EGFRпјҢ6дёӘдёәО”E746-О”E750зјәеӨұпјҢ3дёӘдёәL858Rжӣҝд»ЈгҖӮеҸ‘зҺ°зӘҒеҸҳзҡ„з»Ҷиғһзі»еҜ№ж”ҫе°„зәҝжӣҙж•Ҹж„ҹпјҢеҺҹеӣ жҳҜз»ҶиғһеҜ№еҮәзҺ°зҡ„DNAеҸҢй“ҫж–ӯиЈӮзҡ„дҝ®еӨҚ延иҝҹпјҢеҮӢдәЎеўһеҠ гҖӮиҖҢдё”пјҢеӯҳеңЁеҜ№GefitinibиҖҗиҚҜT790Mзҡ„NSCLCз»Ҷиғһзі»H1975еҗҢж ·еҜ№ж”ҫе°„зәҝж•Ҹж„ҹгҖӮ: ~6 N% f% d- k8 m% [0 \7 j( x
. C3 K5 Z+ ^( t) z6 m9 ]" ?, {8 x дёғгҖҒ NSCLCзҡ„ж–°з”ҹзү©ж Үеҝ—вҖ”вҖ”зҷҢз—Үе№Із»Ҷиғһ" y3 e! {& K3 ]% w! z" G
% v9 q* z1 Y( y3 @5 |5 Y% _ зҷҢз—Үе№Із»ҶиғһжҜ”еҲҶеҢ–зҡ„иӮҝзҳӨз»ҶиғһеҜ№еҢ–з–—жӣҙе…·жҠөжҠ—жҖ§гҖӮзҷҢз—Үе№Із»ҶиғһдёҺе·ІеҲҶеҢ–зҡ„е…¶д»–иӮҝзҳӨз»ҶиғһеҸҜиғҪеӯҳеңЁеҹәеӣ иЎЁиҫҫи°ұзҡ„е·®ејӮпјҢеҰӮжһңиғҪеҸ‘зҺ°иҝҷдәӣе·®ејӮпјҢдёҚд»…дҪңдёәж–°зҡ„з”ҹзү©ж Үеҝ—зү©з”ЁдәҺйў„жөӢиӮҝзҳӨзҡ„з–—ж•ҲпјҢиҝҳеҸҜдҪңдёәжІ»з–—йқ¶зӮ№гҖӮ
$ ~3 ?( a# I& v$ k) y* E" ]: A, H8 q5 {; t @
дҪ“еӨ–з ”з©¶е·Із»ҸеҸ‘зҺ°пјҢд»…д»…1/1000-5000 зҡ„иӮәзҷҢз»ҶиғһиғҪеҪўжҲҗж–°зҡ„е…ӢйҡҶпјҢжҸҗзӨәдёҚжҳҜжҜҸдёӘиӮәзҷҢз»Ҷиғһе…·жңүиӮҝзҳӨиө·жәҗзҡ„еҠҹиғҪгҖӮKimзӯүеҸ‘зҺ°пјҢеҪ“е°Ҹйј жҡҙйңІеҲ°иӮәжҚҹдјӨиҚҜзү©иҗҳдёӯеҗҺпјҢе°‘ж•°иЎЁиҫҫдәҶClara е’ҢиӮәжіЎзү№ејӮж Үеҝ—зҡ„з»ҶиғһеҜ№иҗҳжҳҺжҳҫжҠөжҠ—иҖҢ且继з»ӯеҲҶиЈӮеўһж®–гҖӮиҝҷз§Қз»ҶиғһеҸҜд»Ҙиў«Sca-1pos CD45neg Pecamneg CD34posжүҖиҜҶеҲ«пјҢеңЁдҪ“еӨ–е…·жңүиҮӘжҲ‘жӣҙж–°е’ҢеҲҶеҢ–зҡ„иғҪеҠӣпјҢеҸҜд»ҘеҲҶеҢ–дёәClara з»Ҷиғһе’ҢиӮәжіЎз»ҶиғһпјҢеӣ жӯӨиў«з§°дёәз»Ҷж”Ҝж°”з®ЎиӮәжіЎе№Із»Ҷиғһ(bronchioalveolar stem cellпјҢBASC)гҖӮ) S+ Q- Y: b2 |" X( f2 s# r* [% B
4 f) o# x1 t( f1 T2 ]' @$ ^7 z LSL-K-rasG12Dе°Ҹйј еёҰжңүзҷҢеҹәеӣ K-rasпјҢдҪҶK-rasеӨ„дәҺеӨұжҙ»зҠ¶жҖҒгҖӮеҪ“иҪ¬еҜји…әз—…жҜ’AdCreеҗҺдјҡеҜјиҮҙK-rasиЎЁиҫҫпјҢеҮәзҺ°дёҚе…ёеһӢи…әзҳӨж ·еўһз”ҹ(в…ЎеһӢиӮәжіЎз»Ҷиғһ)пјҢз”ҡиҮіи…әзҳӨе’Ңи…әзҷҢгҖӮеңЁKimзӯүзҡ„з ”з©¶дёӯпјҢйҡҸзқҖиӮҝзҳӨзҡ„еҸ‘з”ҹе’Ңиҝӣеұ•пјҢBASCзҡ„ж•°зӣ®жҳҺжҳҫеўһеҠ гҖӮжҸҗзӨәйҖҡиҝҮжҢҒз»ӯзҡ„K-rasжҙ»еҢ–пјҢеҸҢж Үи®°йҳіжҖ§зҡ„BASCдёәиӮәи…әзҷҢзҡ„иө·жәҗз»ҶиғһпјҢд№ҹе°ұжҳҜиҜҙпјҢиӮәзҷҢе№Із»ҶиғһеҸҜиғҪиө·жәҗдәҺBASCгҖӮ
. J- m/ T8 A5 C0 J2 F4 \& ^$ L3 u/ g/ S
з ”з©¶еҸ‘зҺ°пјҢжҲҗдәәе№Із»ҶиғһиғҪйҖҡиҝҮдҫ§зҫӨ(side populationпјҢSP)иЎЁзҺ°еһӢиҜҶеҲ«вҖ”вҖ”й«ҳжҹ“ж–ҷHoechst 33342жҺ’еҮәиғҪеҠӣгҖӮ2007е№ҙпјҢHungзӯүдҪҝз”ЁжөҒејҸз»ҶиғһжңҜд»Һе…ӯдёӘдәәиӮәзҷҢз»Ҷиғһж ӘдёӯеҲҶзҰ»еҮәSPз»ҶиғһгҖӮ移жӨҚе…Қз–«зјәйҷ·е°Ҹйј е®һйӘҢжҳҫзӨәпјҡдёҺйқһSPз»ҶиғһзӣёжҜ”пјҢSPз»Ҷиғһе…·жңүй«ҳиӮҝзҳӨиө·жәҗзҡ„иғҪеҠӣе’ҢдҫөиўӯиғҪеҠӣгҖӮSPз»Ҷиғһе…·жңүABCG2гҖҒATPз»“еҗҲиҪ¬иҝҗиҪҪдҪ“зҡ„й«ҳиЎЁиҫҫпјҢеҜ№еӨҡз§ҚеҢ–з–—иҚҜзү©иҖҗиҚҜпјӣиҖҢз«ҜзІ’й…¶йҖҶиҪ¬еҪ•иЎЁиҫҫжӣҙй«ҳпјҢжҸҗзӨәе…·жңүж— йҷҗеўһж®–жҪңиғҪгҖӮеҫ®е°Ҹжҹ“иүІдҪ“з»ҙжҢҒиӣӢзҷҪ7(MCM7)зҡ„mRNAж°ҙе№іеңЁSPз»ҶиғһдёӯйҷҚдҪҺпјҢжҸҗзӨәеӨ§еӨҡж•°з»ҶиғһеӨ„дәҺG0йқҷжӯўзҠ¶жҖҒгҖӮеҜ№16дҫӢдёҙеәҠиӮәзҷҢж ·жң¬еҲҶжһҗеҗҢж ·еҸ‘зҺ°еӯҳеңЁзҡ„SPз»ҶиғһгҖӮеӣ жӯӨпјҢиӮәзҷҢдёӯзҡ„SPз»ҶиғһжҳҜиӮәзҷҢиө·жәҗз»Ҷиғһе№Із»ҶиғһпјҢе…¶еҹәеӣ иЎЁиҫҫж Үеҝ—еҸҜиғҪдёҚд»…жҳҜжІ»з–—зҡ„жңүж•Ҳйқ¶зӮ№пјҢд№ҹжҳҜиҚҜзү©ејҖеҸ‘зҡ„дҫқжҚ®гҖӮ! j: q% t/ T# ~3 Q" `
|

 [еӨҚеҲ¶й“ҫжҺҘ]
[еӨҚеҲ¶й“ҫжҺҘ]



 жҜҚдәІжҷҡжңҹиӮәзҷҢи·Ёи¶Ҡ11е№ҙдәҶпјҒжҲ‘们зҡ„5зӮ№
и®Іиҝ°иҖ…пјҡе°ҸзҷҪе…”ж•ҙзҗҶиҖ…пјҡpear
2014е№ҙжҜҚдәІзЎ®иҜҠжҷҡжңҹиӮәи…әзҷҢпјҢдёҖи·Ҝж‘ёзҙўжҠ—зҷҢеүҚиЎҢпјҢе№ёеҫ—з—…еҸӢ
жҜҚдәІжҷҡжңҹиӮәзҷҢи·Ёи¶Ҡ11е№ҙдәҶпјҒжҲ‘们зҡ„5зӮ№
и®Іиҝ°иҖ…пјҡе°ҸзҷҪе…”ж•ҙзҗҶиҖ…пјҡpear
2014е№ҙжҜҚдәІзЎ®иҜҠжҷҡжңҹиӮәи…әзҷҢпјҢдёҖи·Ҝж‘ёзҙўжҠ—зҷҢеүҚиЎҢпјҢе№ёеҫ—з—…еҸӢ
 2026е№ҙе°Ҷжңүе“ӘдәӣиӮәзҷҢж–°иҚҜжңүжңӣзәіе…ҘеҢ»дҝқ
дҪңиҖ…пјҡseacat2025е№ҙеӣҪ家еҢ»дҝқзӣ®еҪ•и°ҲеҲӨеҚіе°ҶејҖе§Ӣз”іжҠҘгҖӮйӮЈдәӣеңЁеӣҪеҶ…иҺ·жү№дёҠеёӮдҪҶжңӘиҝӣе…ҘеҢ»дҝқзӣ®
2026е№ҙе°Ҷжңүе“ӘдәӣиӮәзҷҢж–°иҚҜжңүжңӣзәіе…ҘеҢ»дҝқ
дҪңиҖ…пјҡseacat2025е№ҙеӣҪ家еҢ»дҝқзӣ®еҪ•и°ҲеҲӨеҚіе°ҶејҖе§Ӣз”іжҠҘгҖӮйӮЈдәӣеңЁеӣҪеҶ…иҺ·жү№дёҠеёӮдҪҶжңӘиҝӣе…ҘеҢ»дҝқзӣ®
 23е№ҙи„‘иҶңиҪ¬пјҢзӣ®еүҚи„‘йғЁзЁіе®ҡпјҢиӮәеҺҹеҸ‘зҒ¶
2025.3иҮі2025.5.22и„‘е®һиҙЁе’Ңи„‘иҶңзЁіе®ҡпјҢиӮәйғЁеҺҹеҸ‘зҒ¶еҸҠиғёи…”з§Ҝж¶ІжҺ§еҲ¶дёҚдҪҸгҖӮCEAжҢҒз»ӯеҚҮй«ҳгҖӮ
23е№ҙи„‘иҶңиҪ¬пјҢзӣ®еүҚи„‘йғЁзЁіе®ҡпјҢиӮәеҺҹеҸ‘зҒ¶
2025.3иҮі2025.5.22и„‘е®һиҙЁе’Ңи„‘иҶңзЁіе®ҡпјҢиӮәйғЁеҺҹеҸ‘зҒ¶еҸҠиғёи…”з§Ҝж¶ІжҺ§еҲ¶дёҚдҪҸгҖӮCEAжҢҒз»ӯеҚҮй«ҳгҖӮ
 з”·77е‘ЁеІҒ иӮәи…әзҷҢ 2018е№ҙеҸідёҠиӮәеҲҮйҷӨ
зҲ¶дәІ77еІҒ 2018иӮәи…әзҷҢеҸідёҠиӮәеҲҮйҷӨпјҢд»Ҡе№ҙ6жңҲд»ҪеўһејәCTжЈҖжҹҘжҠҘе‘ҠпјҲеҸідёҠиӮәжңҜеҗҺпјҢжңҜеҢәи§ҒйҮ‘еұһзјқ
з”·77е‘ЁеІҒ иӮәи…әзҷҢ 2018е№ҙеҸідёҠиӮәеҲҮйҷӨ
зҲ¶дәІ77еІҒ 2018иӮәи…әзҷҢеҸідёҠиӮәеҲҮйҷӨпјҢд»Ҡе№ҙ6жңҲд»ҪеўһејәCTжЈҖжҹҘжҠҘе‘ҠпјҲеҸідёҠиӮәжңҜеҗҺпјҢжңҜеҢәи§ҒйҮ‘еұһзјқ
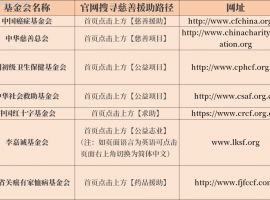 жҠ—зҷҢжҠҘй”Җ80%д»ҘдёҠпјҒеӯҰдјҡиҝҷ5дёӘж–№жі•йғҪиғҪ
дҪңиҖ…пјҡpearжҠ—зҷҢжҳҜдёҖеңәдёҺж—¶й—ҙиөӣи·‘зҡ„жҢҒд№…жҲҳпјҢеҜ№дәҺжӮЈиҖ…家еәӯиҖҢиЁҖпјҢйҷӨдәҶзӣҙйқўз–ҫз—…еёҰжқҘзҡ„жҢ‘жҲҳ
жҠ—зҷҢжҠҘй”Җ80%д»ҘдёҠпјҒеӯҰдјҡиҝҷ5дёӘж–№жі•йғҪиғҪ
дҪңиҖ…пјҡpearжҠ—зҷҢжҳҜдёҖеңәдёҺж—¶й—ҙиөӣи·‘зҡ„жҢҒд№…жҲҳпјҢеҜ№дәҺжӮЈиҖ…家еәӯиҖҢиЁҖпјҢйҷӨдәҶзӣҙйқўз–ҫз—…еёҰжқҘзҡ„жҢ‘жҲҳ



 жҳҫиә«еҚЎ
жҳҫиә«еҚЎ